









July 24, 2026
What Is Application Security as a Service (ASaaS)? A Complete Guide
What Is Application Security as a Service (ASaaS)? A Complete Guide

Omair

Fiza Nadeem is an SEO content writer at ioSENTRIX with 2.5+ years of experience in content writing, copywriting, and SEO.
A 510(k) is a premarket application submitted to the FDA. The name 510(k) comes from the Food, Drug, and Cosmetic Act section it is associated with. This submission is primarily used for Class II devices that aim to prove similar to another device already on the market.
If the FDA determines the device is substantially equivalent, it can be sold immediately without needing a premarket approval (PMA) submission.
The FDA usually does not require clinical data when making a 510(k) submission. However, in some cases, they may ask for clinical data to confirm that the device is substantially equivalent to an existing one. This doesn't mean that you should not consider collecting clinical data for other reasons, such as getting a refund or promoting the adoption of your device.
Preparing a 510(k) can be complex, and the FDA has strict guidelines for the submission process. While some companies manage the process independently, expert guidance often improves outcomes.
Even though it is considered one of the "easier" submissions, a 510(k) can exceed 100 pages and include many information sections. It also needs to comply with the FDA's electronic document submission requirements.
Manufacturers looking to bring Class II medical devices and the Class I and unclassified devices mentioned earlier to the U.S. market must submit a 510(k) to the FDA. Additionally, a 510(k) is necessary for manufacturers who alter the intended use of their medical device or change the technology of an already cleared device in a way that could significantly impact its safety or effectiveness, even if the change is an improvement.
A medical device must fulfill specific requirements for 510(k) clearance. We have outlined and summarized these criteria below:
Note: The list provided is not complete. Other factors must be considered based on the regulatory landscape and the specific device. Medical device companies often work with regulatory consultants with expertise in this area to manage these complexities.
An experienced consultant can assist in determining whether a device qualifies for 510(k) clearance and help ensure that the submission complies with all regulatory standards.
To ensure both safety and efficiency, the FDA has established three levels of oversight based on the risk of each device. The first level covers low-risk devices, which do not require premarket submission. The second level, designed for medium-risk devices, is known as Premarket Notification or 510(k).The third level, which applies to high-risk devices, is known as Premarket Approval (PMA).
Some types of devices cannot undergo the 510(k) clearance process. These include devices meant to be part of another device, custom-made devices, and those intended for investigational use.
The device should not have a past record of being banned, withdrawn, or restricted by the FDA. It must also not have been previously denied for 510(k) clearance or premarket approval.
The device should not present an unreasonable risk to public health or safety and needs to be produced according to the 21 CFR 820 Quality System Regulation (QSR). It must also comply with relevant performance standards set by the FDA or international standards organizations.
A successful 510(k) requires meeting several essential criteria. Some of these requirements are clear and directly relate to the earlier eligibility standards. However, others may not be as obvious so you may need the assistance of an experienced regulatory consultant. This expert can clearly understand what the FDA expects and can prepare the necessary documentation in detail from the very beginning.
Medical device manufacturers need to identify their device's correct classification and understand the related regulatory requirements. This information is essential for deciding on the right 510(k) submission type to file.
Complete documentation of the device's design and function is essential for the 510(k) submission. This documentation must include drawings, schematics, and a clear explanation of the device's intended use and performance.
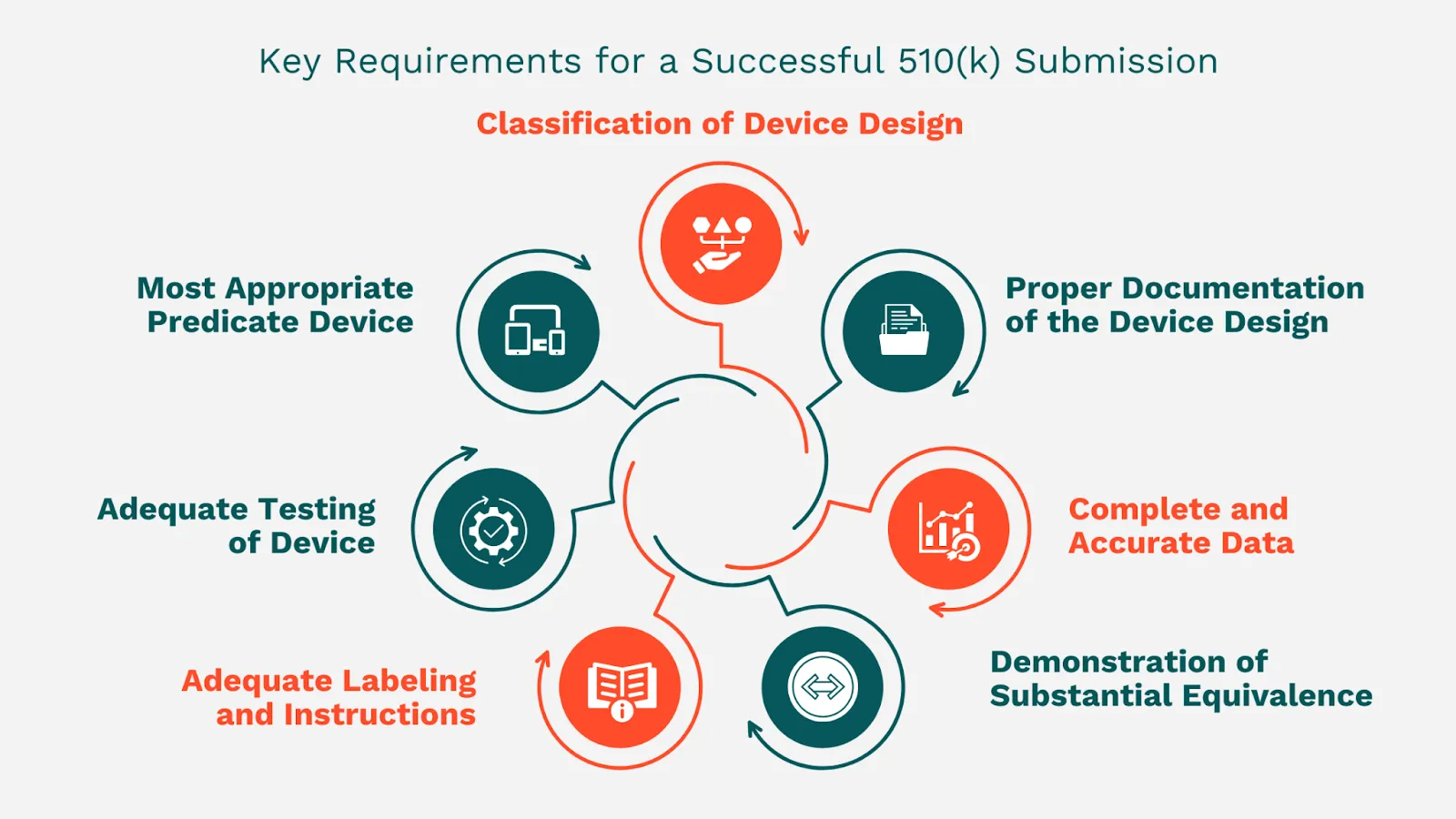
The 510(k) submission must compare between the new device and a similar device already on the market, known as a predicate device. The chosen predicate device should be alike in design, intended use, and technology.
The 510(k) submission must prove substantial equivalence to the predicate device and does not introduce new concerns regarding safety and effectiveness.
The labeling and instructions must be thorough and precise, and they must offer enough information to ensure the user's safe use of the device.
Sufficient testing must show that the device meets all relevant standards. The testing should be thorough and suitable for the type of device and its intended use.
The data in the 510(k) submission must be thorough, correct, and well-structured. The FDA will not accept incomplete submissions or submissions with false or misleading information.
This is not a complete list, but a few examples of issues that can be prevented by collaborating with the right regulatory consultant.
This issue can affect all companies, but startups, smaller firms, and new medical device manufacturers may not fully grasp the key details of the process. Therefore, getting expert advice from an experienced regulatory professional is essential.
As mentioned earlier, to qualify for clearance through this regulatory route, a device must show that it is substantially equivalent to an already approved device, referred to as a predicate device. This can often be challenging, as some manufacturers have difficulty identifying a suitable predicate device.

The 510(k) submission must include thorough and organized documentation that proves the device's safety and effectiveness. This documentation should contain data about the device's design, materials, and manufacturing processes, among other things. Companies may think they have provided enough information, but the FDA often does not consider this the case.
The FDA is likely to have questions or concerns regarding the device, so it is essential for the manufacturer to anticipate these and offer clear and convincing answers. Without the assistance of an experienced regulatory consultant, it can be challenging to predict what these questions might be and to respond to them effectively.
A common mistake medical device companies make when submitting a 510(k) application is failing to communicate effectively with the FDA reviewers. Engineers working on the device often think that FDA reviewers have the same technical knowledge, which is often not the case.
Although FDA reviewers may have a general understanding, they typically do not have the specific technical expertise about your device that your engineering team has. Therefore, it is important to offer a clear and simple explanation of the device that can be understood by everyone, including those who are not as familiar with it.
If you can remember only three key points from this blog, keep these in mind:
Identify and define a predicate for your medical device before submitting your 510(k) application. If you do not have a suitable predicate, your device will not be cleared for market entry through this process, which can result in delays and higher costs.
Small businesses need to pay close attention to FDA fees and timelines, as well as the costs of lab testing. These elements can have a major effect on their revenue.
Delays are likely to occur if there is no explicit predicate and the submission is not well-prepared. For a successful submission, understand the timeline and be ready for possible challenges.




